Testing out Seqan's Multipattern Search Implementations
I recently discovered Seqan, a header-only C++ library for bioinformatics. I’ve been playing around with the toolkit to make some small programs just to see whether I want to use it in a larger project. So far I’ve written prepmate, an adaptor trimming program for Illumina’s Nextera mate-pair libraries; and fxtract, a grep-like program for extracting fasta/fastq records from large files. One of the algorithms that I use in fxtract and in another program I’ve written, crass, is to search for multiple patterns simultaneously (in this case a number of different DNA motifs). Seqan implements a number of algorithms for multipattern matching (checkout their tutorial page), however they don’t give many clues as to why you would choose one algorithm over another. So I decided to take a few of these implementations out for a spin using fxtract.
Test variables:
- different numbers of patterns: 100, 1000, 10000
- different length patterns: uniformly distributed size (0 - 100bp), 30bp, 60bp
- Algorithms: WuManber, MultiBfam, AhoChorasick, SetHorspool
- Test dataset: SRR438796, which is part of an EBPR metagenome that was sequenced with Illumina GAIIx
I should point out the the following speed tests are not necessarily testing the algorithms, but the seqan implementation of these algorithms in the context of what fxtract is trying to accomplish. Which means that other programs which implement these algorithms may perform quite differently and the conclusions that I reach below may be different in other situations. So with disclamers out of the way, how does each implementation perform.
While running through a test of the wumanber implementation I discovered something very odd when using the uniformly distributed patterns. With 100 patterns the program ran through fine, outputting the correct results, however when using the 1000 or 10000 pattern file I got no output. I discovered though that these larger files contained some patterns that had a length of 0 (i.e. they were empty lines), whereas the 100 pattern file’s smallest pattern was 6bp. I removed the empty lines from my pattern files and then and voila there was output. What this means is that the seqan WuManber implementation fails silently when given an empty string as one of the patterns! I’ve submitted a bug report with the seqan authors.
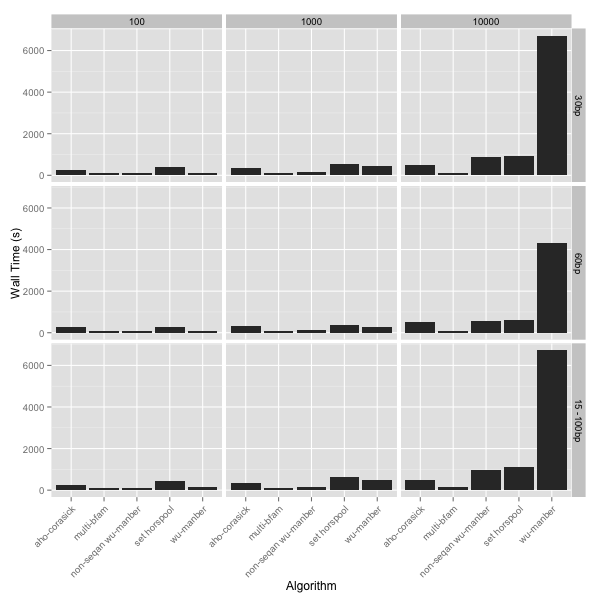
Seqan speed test summary
The above graph shows the total wall clock time in seconds for each
algorithm to process the input file with the different numbers of search
patterns (columns) or for the different types of patters (rows).
The main thing that striked me was the poor performance of the
seqan implementation of the Wu-Manber algorithm - it does not scale well.
I was lead to believe that Wu-Manber was the pinicle of multipattern searching so I was a little perplexed by this result. I know of another stand alone implementation of Wu-Manber provided by Ray Burkholder. As a comparison I tested out this implementation as well and perhaps unsurprisingly this implementation performed much better (it’s referred to in the figures as “non-seqan wu-manber”). Bottom line: So don’t use the Wu-Manber implementation in Seqan! The winner of the speed benchmark was the multiBfam algorithim which performed nearly identically for any number of input patterns or for the different length patterns. In comparison the Aho-Corasick and SetHorspool algorithms became slower with more patterns.
Speed tells you only half of the story and usually comes at the price of memory consumption. Both of the Wu-Manber implementations excelled at memory consumption, or lack there of. The worst were the Aho-Corasick and SetHorspool algorithms, which used 20Gb+ of memory under the worst case. The multiBfam algorithm was somewhere in the middle (~7Gb under the worst case scenario).
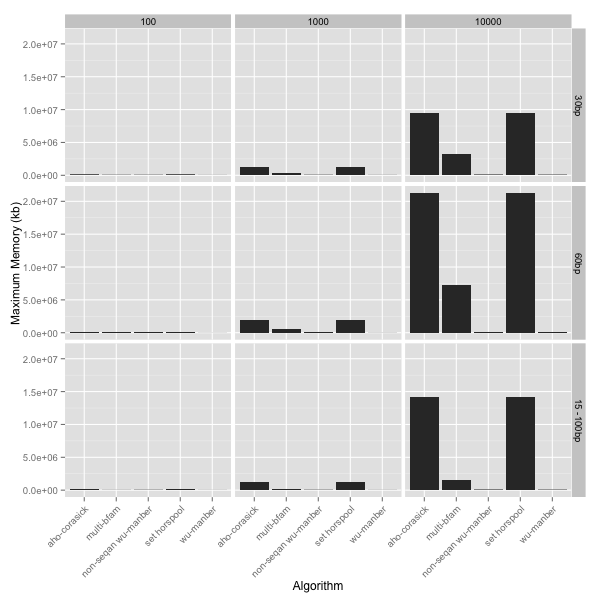
Seqan speed test summary
Considering only the seqan implementations I would probably go for the
multiBfam algorithm as my first choice when searching for less than 1000
patterns after which Wu-Manber would have to be preferred.
Base on what I’ve seen the Aho-Corasick and SetHorspool implementations
in seqan are the worst options as they use far too much memory, however
that’s not to say that other implementations of these algorithms would
perform differently.
Scripts
make_patterns.sh
Running this script requires that both seqtk and bioawk are installed and in your PATH.
for count in 100 1000 10000
do
# make uniformly distributed patterns
seqtk sample SRR438796.fastq.gz
$count |\
bioawk -c fastx '{
start=rand() *99;
end=rand() * 99;
if(start < end){
if(end - start > 15) {
print substr($seq, start, end)
}
}else{
if(start - end > 15) {
print substr($seq, end, start)
}
}
}' >random_${count}_patterns.txt
# make 30bp patterns
seqtk sample SRR438796.fastq.gz
$count |\
bioawk -c fastx '{
start=rand() * 30;
end=start + 30;
print substr($seq, start, end)
}' >random_${count}_30bp_patterns.txt
# make 60bp patterns
seqtk sample SRR438796.fastq.gz
$count |\
bioawk -c fastx '{
start=rand() * 30;
end=start + 60;
print substr($seq, start, end)
}' >random_${count}_60bp_patterns.txt
done
####time_implementations.sh
#! /bin/bash
for impl in fxtract-*
do
for p in speed_test/random_100*
do
patterns=$(basename $p)
echo "real,user,sys,avemem,maxmem" >speed_test/${impl}_${patterns%.txt}_times.txt
for i in {1..10}
do
/usr/bin/time -f "%E,%U,%S,%K,%M" -a -o speed_test/${impl}_${patterns%.txt}_times.txt ./$impl -f $p SRR438796.fastq.gz |\
wc -l >>speed_test/${impl}_${patterns%.txt}_words.txt
done
done
done
summarize_times.py
#!/usr/bin/env python
from __future__ import print_function
import sys, os, glob, re
fixed_re = re.compile('fxtract-(\w+)_random_(10{2,5})_(\d\d)bp_patterns_times\.txt')
norm_re = re.compile('fxtract-(\w+)_random_(10{2,5})_patterns_minus_small_times\.txt')
impl_codes = {'ac': 'aho-corasick', 'sh': 'set horspool', 'mb': 'multi-bfam', 'wm': 'wu-manber'}
times_files = glob.glob("./fxtract*times.txt")
data = {}
for fn in times_files:
match = fixed_re.search(fn)
if match:
impl = impl_codes[match.group(1)]
pattern_type = match.group(3)
pattern_count = match.group(2)
else:
match = norm_re.search(fn)
if match:
impl = impl_codes[match.group(1)]
pattern_type = 'norm'
pattern_count = match.group(2)
else:
raise ValueError("Filename %s does not fit either form" % fn)
with open(fn) as fp:
first_line = True
seconds = 0.0
for line in fp:
if first_line:
first_line = False
continue
fields = line.rstrip().split(',')
seconds += float(fields[1]) + float(fields[2])
seconds /= 10.0
print(impl,pattern_type, pattern_count, seconds, sep="\t")
plot_summary.R
library(ggplot2)
data = read.table("summary.txt", header=T, sep="\t")
p <- ggplot(data, aes(Algorithm, Wall.time)) + geom_bar(stat="bin")
png("seqan_multipattern_search_speed_benchmark.png", width=600,
height=600)
p + facet_grid(Pattern.type ~ Number.of.patterns) + theme(axis.text.x =
element_text(angle = 45, hjust = 1))
dev.off()